

What Is Flow Cytometry?
Complete the form below to unlock access to ALL audio articles.
Contents
What are forward scatter, side scatter and fluorescent signals?
What instruments are used in flow cytometry?
- Developments in flow cytometry instrumentation
- Cell sorting
How are flow cytometry data analyzed?
- Selection of viable single cells
- Compensation
- Gating and analysis
- Analysis of high-dimensional data
What are the applications of flow cytometry?
Challenges and limitations of flow cytometry
When it comes to analyzing cells in a lab setting, flow cytometry is a widely used and comprehensive single-cell analysis method. This article outlines what flow cytometry is, how it works, the different types that exist, how data are analyzed, and what the future holds for flow cytometry.
What is flow cytometry?
Flow cytometry is a technique used to analyze cells for a variety of purposes, including cell counting, phenotyping, cell cycle assessment, and viability. The light produced by lasers in a flow cytometer is scattered by cells in the sample, measured by detectors, and then translated to signals that can be analyzed and measured.
How does flow cytometry work?
To get started with flow cytometry, the first step is to prepare the sample to be analyzed. Cells obtained from cell culture, blood, or disaggregated tissues, should be made into a suspension. This cell suspension is divided over several tubes for staining, retaining an aliquot of unstained cells as a control. The other samples are stained by adding antibodies labeled with fluorescent probes, or dyes that stain cellular components. When analyzing intracellular proteins, the cells first must be fixed (in a formalin buffer) and permeabilized (with a permeabilizing agent) to allow antibodies and dyes to enter the cell. After incubation with the antibodies or dyes, the cells are washed in buffers and resuspended in a saline-based buffer for analysis. This prepared sample is then introduced to the flow cytometer.
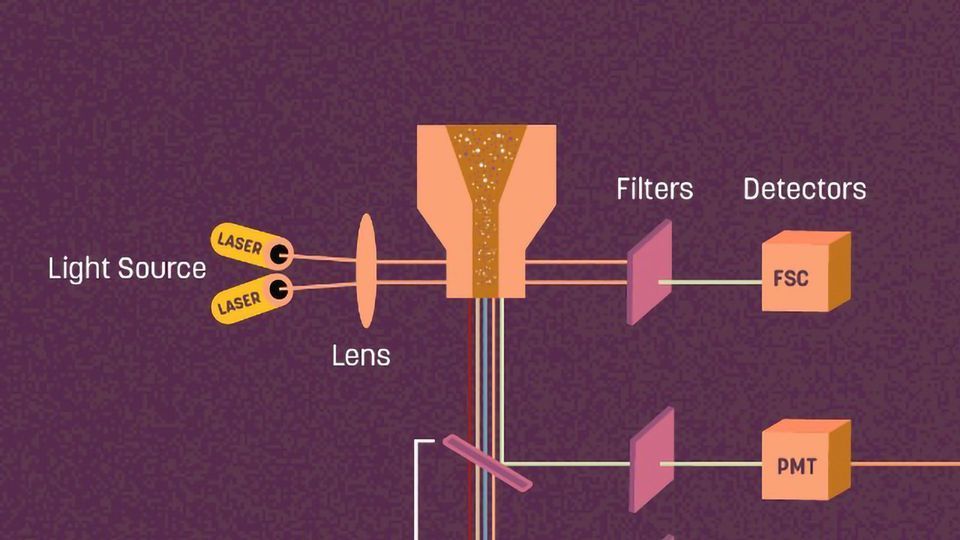
There are three main components in a flow cytometer:1 fluidics, optics, and electronics. In the central part of the cytometer - the flow cell - the sample material meets the excitation light and scatters light that is subsequently picked up by the detection systems.  Figure 1: The main flow cytometry components. Credit: Technology Networks.
Figure 1: The main flow cytometry components. Credit: Technology Networks.
1. The fluidics system contains sheath fluid (a saline-based buffer or water) that is pressured through the machine to guide the cell sample past the laser for separate measurement of every single cell.
2. The optics consist of lasers, which emit light to the samples, and the collection optics, such as photomultiplier tubes (PMTs) that collect the signal scattered by the sample.
3. Finally, the cytometer's electronics convert the detected signal to digital parameters that can be analyzed using software.
What are forward scatter, side scatter and fluorescent signals?
The three primary output measurements of flow cytometry are forward scatter, side scatter, and fluorescent signals. The laser's visible light reflects off each cell providing the general size and shape characteristics of the cell. Figure 2: Forward scatter. Credit: Technology Networks.
Figure 2: Forward scatter. Credit: Technology Networks.The forward scatter (FSC) is the scatter coming from the forward direction and reflects the cells' size. The scatter is measured along the path of the laser and each cell will bend the light around the sides of the cell causing diffraction. Therefore, the intensity of FSC reflects the diameter of the cell.
 Figure 3: Side scatter. Credit: Technology Networks.
Figure 3: Side scatter. Credit: Technology Networks.The side scatter (SSC) is the scatter measured at 90 degrees of the laser beam and reflects the granularity or the complexity of the cell. The structures in the cell will change the direction of light waves that enter the cell, resulting in specific cellular structures, such as granules and the nucleus, to refract the light. The more intense the SSC, the more refraction, the more granularity is expected in the cell.
 Figure 4: Fluorescent signal. Credit: Technology Networks.
Figure 4: Fluorescent signal. Credit: Technology Networks.The third output measure is the fluorescent signal. This the light that is emitted when the laser excites fluorophores. Fluorophores are molecules that can absorb and re-emit light upon excitation. Photons from the laser light source are absorbed by the fluorophore’s electrons, moving them to a higher energy state. These electrons then release energy as photons to return to their ground state. The wavelength of these photons is impacted by the amount of energy lost to molecular interactions during the process. Each fluorophore has its own emission spectrum which may partially overlap with that of other fluorophores. Fluorophores can be used to label antibodies that in turn can bind to and thus detect specific antigens on or in a cell, or can be used as a dye to stain cells directly, for instance, by binding to DNA.
What instruments are used in flow cytometry?
At a minimum, a cytometer will contain a laser, a detector, and a fluidics system, ensuring the fast flow of cells past the laser beams. The number of lasers in a cytometer and the number and quality of detectors has significantly increased over the years. These developments have expanded the number of markers that can be detected at once.Flow cytometers may be designed for specific purposes. For example, to allow a high throughput of samples, well plate loaders may be incorporated to enable quick hands-off analysis of multiple samples.
Developments in flow cytometry instrumentation
To add to the multiparameter analyses and allow for more in-depth assessments, other types of cytometers have been developed:Mass cytometry2 (time-of-flight mass spectrometry or cyTOF): Instead of antibodies labeled with fluorophores, this technique uses antibodies labeled with heavy metal ion tags. Over 100 metal probes are available with limited signal overlap, allowing for more high-dimensional analysis than traditional flow cytometry. Therefore, in-depth phenotyping can be performed with this technique, useful in research areas such as immunology and cancer. The main difference with traditional flow cytometry is that the droplets containing cells for analysis are vaporized, atomized, and ionized in the flow chamber, and the mass spectrometer detects the ions produced. The time-of-flight of the ions from the sample to the detector is used for the analysis.
Spectral flow cytometry3: Rather than assessing each fluorophore's peak emission, spectral flow cytometry uses a series of detectors to measure the entire spectrum of each fluorophore. These spectra are then separated using algorithms. This allows for the use of fluorophores with overlapping spectra to increase the number of parameters analyzed in one experiment.
Imaging cytometry4: This type of cytometry combines traditional flow with fluorescence microscopy. The technique additionally allows for the assessment of the morphology of the cells and can be used to visualize co-expression of proteins, cell binding, nuclear translocation of proteins and immune cell synapses.
Cell sorting
A fluorescence-activated cell sorter (FACS) is a device that uses flow cytometry to sort cells for further analysis or experiments1. Instead of only measuring cell characteristics, the cells are selected based on specific characteristics and guided into collection vessels to purify the sample for certain cell types. An example of the use of FACS is to obtain T cells from a sample of peripheral blood mononuclear cells (PBMCs) from blood. As further culturing of cells is often required after sorting, it is essential that sorting is performed in sterile conditions and that the procedure is gentle to avoid damaging the cells and reducing their viability. For this reason, cell sorting cannot be performed in mass spectrometry due to the destruction of cells for analysis. Sorters can be traditional flow cytometry-based or spectral-based.Sorting is performed on cell populations that are positive or negative for a particular parameter. Cells are separated using high-frequency oscillation of the single cell sample liquid stream to create drops that are given a positive or negative charge. The droplets are then directed to a collection vessel based on their charge.
How are flow cytometry data analyzed?
Histograms and dot plots are created from the flow cytometry data collected to allow analysis. But first, there are several processing steps that should be taken to enable accurate analysis.Selection of viable single cells
Generally, an analysis will start with the selection of single cells of interest based on certain characteristics, which is done by a process known as gating. Analysis software allows users to draw lines, known as gates, around certain cell populations to select them for further analysis. It is essential to gate for single cells as doublets, cells that pass the laser simultaneously, can result in false-positive signals from one of the cells. Using dot plots showing the FSC and SSC data, single cells with the size and granularity of a population of interest are selected for further analysis.Additionally, it is essential to gate for viability, as dead cells tend to emit more autofluorescence which may affect analysis. Autofluorescence may overlap with some of the fluorophores used resulting in false positive signals. Viability is assessed using viability dyes, which can be DNA-binding or react with free amine groups on and in the cells.
Compensation
The next step for analysis is the assessment of the specific fluorescent signals measured for each cell. As there is overlap in the emission spectra of available fluorophores, experiments with multiple fluorophores may require compensation.5 Overlapping spectra can lead to a false positive, as the measured signal is coming from another fluorophore. Therefore, proper experimental design is essential to construct fluorophore panels with minimal spectral overlap. Even then, correction for overlap, or compensation, is often required. A positive and negative control sample guides the compensation, ideally using the studied cell type, or if this is not possible, using compensation beads.5 Compensation can be set during the measurement or in the analysis afterward.Gating and analysis
Once compensation is set, each marker can be analyzed by gathering data from each fluorescent detection channel. Additional gating can be performed on populations of interest to focus on the presence or absence of specific parameters. Both density and histogram plots can be used for this purpose.16080258934381625750565937.jpg "Figure 5: Representation of a density plot and histogram.") Figure 5: Representation of a density plot and histogram. Credit: Technology Networks.
Figure 5: Representation of a density plot and histogram. Credit: Technology Networks.
Density plots show individual cells as a dot distributed according to their fluorescence properties. Colors are used to indicate the number of cells in the analysis with those characteristics. Two markers can be visualized at once, one on the X-axis and one on the Y-axis, differentiating cells based on the expression of these two parameters. Histograms display a single parameter and show the cell counts on the Y-axis and the fluorescence intensity on the X-axis.
Controls are essential for proper gating, which can be set based on positive and negative controls. A positive control is a cell sample expressing the marker detected by an antibody with the fluorophore of interest. A negative control could be unstained cells or cells stained with an isotype control; an antibody labeled with the fluorophore used in analysis which is targeted against an antigen not present on the cell, to correct for non-specific background. For bigger panels with many parameters, it is recommended to use fluorescence-minus-one (FMO) controls. These controls include all fluorescent markers of an individual flow panel minus one and should be created for every fluorescent marker used in the experiment. It allows for the detection of background signals from overlapping spectral profiles.
Analysis of high-dimensional data
The expansion of the number of parameters studied has changed the way flow cytometry data is analyzed. With each parameter added, the data gains more dimensions. Therefore, new analysis techniques have been developed to extract information from these high-dimensional data. Examples include principal component analysis (PCA),6 spanning-tree progression analysis of density-normalized events (SPADE),7 and t-stochastic neighbor embedding (tSNE) algorithms.8 These algorithms cluster data based on phenotypic similarities and differences between cells to allow two-dimensional analysis.What are the applications of flow cytometry?
Flow cytometry is used for cell biology, including during biopharmaceutical development and analysis, and in more clinical applications.Cell biology applications include:
A) Cell counting
B) Analysis of the cell cycle
C) Measuring viability
D) Proliferation
E) Phenotyping of various cell types and subsequent cell sorting

For (pre)clinical applications flow cytometry can be used to:
A) Detect various immune cells9
B) Assess cytokine production upon activation of immune cells and their potential reactivity towards target cells10
C) Study cancer cells and cells in the tumor microenvironment in-depth11
D) Detect virus-infected cells12
E) Assess the characteristics of spermatozoa in fertility analyses13
F) Monitor cardiac disease and sepsis14,15
G) Characterize cells in neuroscience research16
Challenges and limitations of flow cytometry
Despite the advantages that flow cytometry can offer to cell biology and clinical applications, there are some downsides associated with the technique. The fluidics in the system can be challenging to maintain consistent, with cells or debris causing blockades or changes in the flow rates that affect analysis. There is particular risk for this when larger cells, such as cancer cells, are analyzed. Contamination in the piping can cause the system to get blocked and flow to be interrupted. Laser alignment is also essential to ensure consistent results and should be checked regularly. Additional limitations include the potential of the cytometer to cause damage to cells, affecting the analysis and subsequent culturing when sorting cells, and the inability of single cell analysis to provide information on tissue characteristics. Finally, the costs of cytometers and the operating costs, in particular for high parameter devices, can be high, limiting its use.Future developments in flow cytometry
Over the past decades, the main developments in the field have allowed for significant increases in the number of parameters that can be studied in each sample and a reduction in the size of these bulky machines. These developments are likely to continue over the coming years, only increasing the complexity of obtained data, with a need to develop software to standardize and validate such analyses. Further developments in this area will allow for more extensive analyses on a broader scale, increasing the number of markers that can be analyzed for each sample and advancing data analysis methods to allow large sample sizes to be analyzed in detail. Reductions in the instruments' sizes will allow for easier placement on the benchtop of labs and might even enable use in limited formats in the field. Additionally, more standardization of workflow will reduce user error rates and allow high throughput clinical and diagnostic applications. Automatic pipetting, setting of compensation, and allowing well plates to be analyzed automatically can increase the reliability of flow cytometry analyses.
Glossary
| Antibody | Protein that specifically binds an antigen, which can be labeled with fluorophores or other chemicals for detection |
| Autofluorescence | Natural emission of light by cells |
| Compensation | Calculation to remove fluorescent spectral overlap spilling over in multiple detectors |
| Compensation beads
| Microparticles that easily bind antibodies, which can serve as a positive control to determine the right settings for the flow cytometer |
| Density plots | A plot in which the distribution of cells with certain fluorescence intensity is provided with colors reflecting the density of cells with those characteristics |
| Dot plots | Plot in which each cell is represented as a dot |
| Doublets | When two particles or cells pass through the laser light simultaneously |
| FACS | Fluorescence activated cell sorter |
| Fluorophore | Or fluorochrome, a fluorescent chemical which emits light after laser excitation |
| FMO | Fluorescence minus one – specific control, leaving out one of the fluorophores of the complete panel to account for spillover |
| Forward scatter | FSC; scatter coming from the forward direction of the cell reflecting the cells' size |
| Gating | Process of selecting cell populations of interest based on cellular characteristics |
| Histogram | Graph depicting the fluorescence or light scatter intensity with the number of cells on the y-axis |
| Isotype control | Antibody raised against an antigen not found on the cell of analysis, used to assess non-specific binding |
| Laser | Emits light at a specific wavelength |
| PBMCs | Peripheral blood mononuclear cells, peripheral blood cells consisting of lymphocytes and monocytes |
| Photomultiplier tube | PMT, a sensitive detector of light at various ranges of the electromagnetic spectrum |
| Side scatter | SSC; scatter measured at 90 degrees of the laser beam reflecting the granularity or the complexity of the cell |
| Spillover | Fluorescent signal of one fluorophore is measured in the channel of another fluorophore, when both are measured on the same cell, resulting in a false positive signal |
References

